Apert Syndrome
Updated: Sep 2, 2009
Information from Industry
Clinical studies in treating eczema with colloidal oatmeal- based skincare
Over-the-counter, colloidal oatmeal-based skincare regimen proven in 2 clinical studies in eczema.
Review clinical trial data
Over-the-counter, colloidal oatmeal-based skincare regimen proven in 2 clinical studies in eczema.
Review clinical trial data
Introduction
Background
- Apert syndrome is named for the French physician who described the syndrome acrocephalosyndactylia in 1906.
- Apert syndrome is a rare autosomal dominant disorder characterized by craniosynostosis, craniofacial anomalies, and severe symmetrical syndactyly (cutaneous and bony fusion) of the hands and feet.
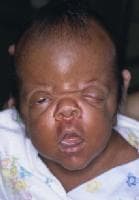
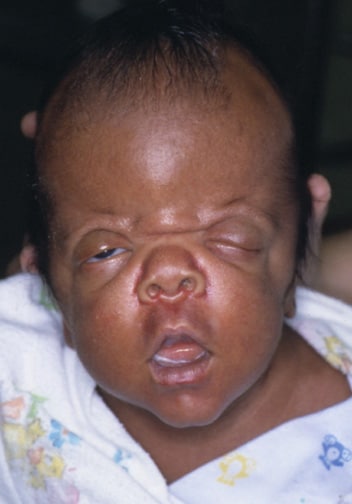
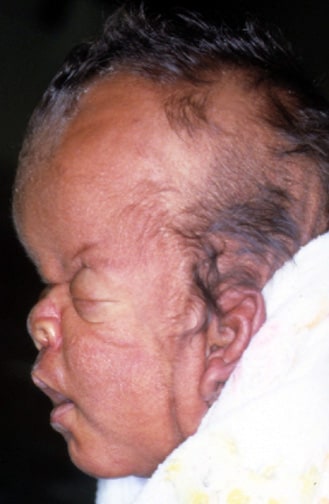
An infant with Apert syndrome is shown. Note the characteristic ocular hypertelorism, down-slanting palpebral fissures, proptotic eyes, horizontal groove above the supraorbital ridge, break of the continuity of eyebrows, depressed nasal bridge, and short wide nose with bulbous tip.
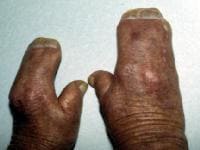
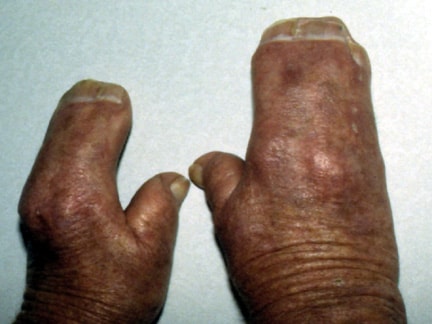
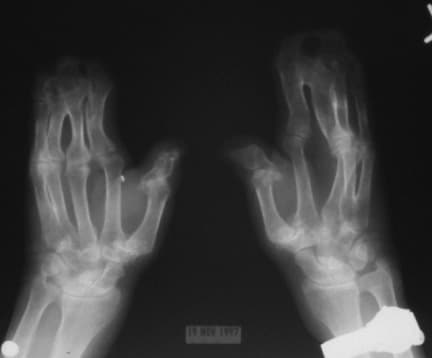
Note the mitten appearance of the hands with syndactyly involving the second, third, fourth, and fifth fingers. This patient also has characteristic concave palms, hitchhiker posture (radial deviation) of short broad thumbs, and contiguous nailbeds (synonychia).
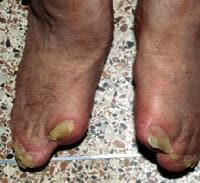
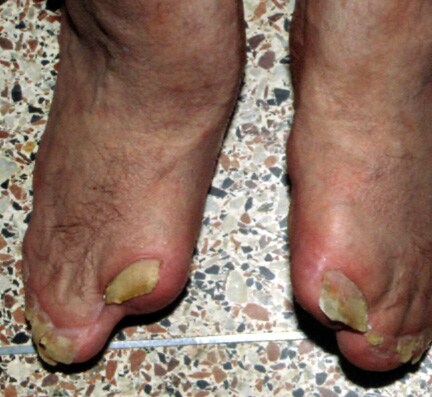
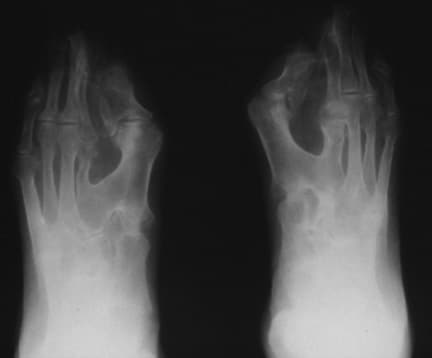
Note the sock appearance of the feet with syndactyly involving the second, third, fourth, and fifth toes. The patient also has contiguous nail beds (synonychia).
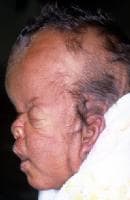
In this profile, turribrachycephaly, high prominent forehead, proptosis, depressed nasal bridge, short nose, and low-set ears are prominent.
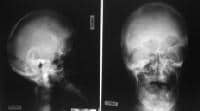
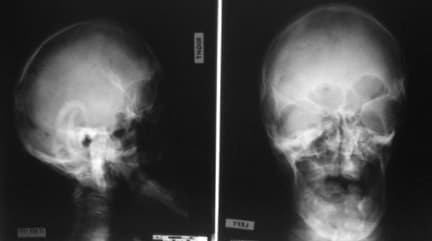
This radiograph demonstrates turribrachycephaly, shallow orbits, ocular hypertelorism, and hypoplastic maxilla.
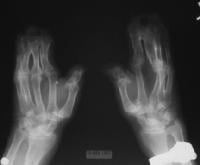
Note osseous syndactyly involving the second, third, fourth, and fifth fingers; multiple synostosis involving distal phalanges and proximal fourth and fifth metacarpals; symphalangism of interphalangeal joints; shortening and radial deviation of distal phalanx; and delta-shaped deformity of proximal phalanx of the thumbs.
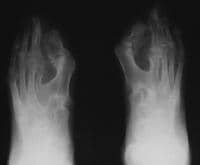
Note osseous syndactyly, fusion of interphalangeal joints, synostosis involving proximal first and second metatarsals, and partially duplicated and delta-shaped proximal phalanx of the great toes.
- It is probably the most familiar and best-described type of acrocephalosyndactyly.
- Reproductive fitness is low, and more than 98% of cases arise by new mutation.
Pathophysiology
- During early infancy (<3 mo), the coronal suture area is prematurely closed. A bony condensation line beginning at the cranial base and extending upward with a characteristic posterior convexity represents this occurrence. Anterior and posterior fontanelles are widely patent. The midline of the calvaria has a gaping defect, extending from the glabellar area to the posterior fontanelle via the metopic suture area, anterior fontanelle, and sagittal suture area. The skull with a gaping midline defect appears to permit adequate accommodation of the growing brain. The lambdoidal sutures appear normal in all cases.
- During the first 2-4 years of life, the midline defect is obliterated by coalescence of the enlarging bony islands without evidence of any proper formation of sutures. An extreme short squama and orbital part of the frontal bone together with the posterior convexity of the coronal bone condensation line suggest that growth inhibition in the sphenofrontal and coronal suture area has its onset very early in fetal life.
- Unique fibroblast growth factor receptor 2 (FGFR2) mutations lead to an increase in the number of precursor cells that enter the osteogenic pathway. Ultimately, this leads to increased subperiosteal bone matrix formation and premature calvaria ossification during fetal development. The order and rate of suture fusion determine the degree of deformity and disability. Once a suture becomes fused, growth perpendicular to that suture becomes restricted, and the fused bones act as a single bony structure. Compensatory growth occurs at the remaining open sutures to allow continued brain growth; however, complex, multiple sutural synostosis frequently extends to premature fusion of the sutures at the base of the skull, causing midfacial hypoplasia, shallow orbits, a foreshortened nasal dorsum, maxillary hypoplasia, and occasional upper airway obstruction.
- The first genetic evidence that syndactyly in Apert syndrome is a keratinocyte growth factor receptor (KGFR)-mediated effect was provided by the observation of the correlation between KGFR expression in fibroblasts and severity of syndactyly. Patients with Ser252Trp and those with Pro253Arg have different phenotypic expression. The syndactyly is more severe with Pro253Arg mutation for both hands and feet, whereas cleft palate is significantly more common with Ser252Trp mutation.1
- Amblyopia and strabismus is more common in patients with the FGFR2 Ser252Trp mutation, and optic disc pallor is more frequent in patients with the FGFR2 Pro253Arg mutation.2 Patients with FGR2 Ser252Trp mutations have a significantly greater prevalence of visual impairment compared with patients with the FGFR2 Pro253Arg mutation.3,4
Frequency
United States
- Prevalence is estimated at 1 in 65,000 (approximately 15.5 in 1,000,000) live births.5,6,7
- Apert syndrome accounts for 4.5% of all cases of craniosynostosis.
Mortality/Morbidity
- Most patients experience some degree of upper airway obstruction during infancy. Upper airway compromise due to reduction in nasopharynx size and choanal patency as well as lower airway compromise due to anomalies of the tracheal cartilage may be responsible for early death.
- Sleep apnea syndrome is common. Upper airway compromise, consisting of obstructive sleep apnea and cor pulmonale, may result from small nasopharyngeal and oropharyngeal dimension in the Apert craniofacial configuration.
- Patients are at risk for complications resulting from elevated intracranial pressure despite surgical attempts to increase cranial capacity in infancy.
Race
- Asians have the highest prevalence (22.3 cases per million live births).
- Hispanics have the lowest prevalence (7.6 cases per million live births).
Sex
- Apert syndrome has no sex predilection.
Age
- Apert syndrome is detected in the newborn period due to craniosynostosis and associated findings of syndactyly in the hands and feet.
Clinical
History
- Family history is usually not significant because most cases of Apert syndrome are sporadic. A paternal age effect increases in fathers older than 50 years.
- Headache and vomiting are signs of acute increased intracranial pressure, especially in cases of multiple suture involvement.
- Stridor and sleep apnea indicate problems with the upper airway, resulting from craniosynostosis of sutures of the base of the skull.
- Visual disturbance can result from corneal injury due to exposed conjunctivitis and keratitis.
- Many patients exhibit mental retardation, although patients with normal intelligence have been reported.
Physical
- Skull and face
- Craniostenosis is present. Coronal sutures most commonly are involved, resulting in acrocephaly, brachycephaly, turribrachycephaly, flat occiput, and high prominent forehead.
- Large late-closing fontanels are observed.
- A gaping midline defect is present.
- A rare cloverleaf skull anomaly is present in approximately 4% of infants.
- Common facial features during infancy include horizontal grooves above the supraorbital ridges that disappear with age, a break in the continuity of the eyebrows, and a trapezoid-shaped mouth at rest.
- A flattened, often asymmetric face is observed.
- Maxillary hypoplasia with retruded midface is present.
- Ears, eyes, nose, and mouth
- Patients have apparent low-set ears with occasional conductive hearing loss and congenital fixation of stapedial footplate.
- Eyes exhibit down-slanting palpebral fissures, hypertelorism, shallow orbits, proptosis, exophthalmos, strabismus, amblyopia, optic atrophy, and, rarely, luxation of the eye globes, keratoconus, ectopic lentis, congenital glaucoma, lack of pigment in the fundi with occasional papilledema, and preventable visual loss or blindness.
- The nose has a markedly depressed nasal bridge. It is short and wide with a bulbous tip, parrot-beaked appearance, and choanal stenosis or atresia.
- The mouth area has a prominent mandible, down-turned corners, high arched palate, bifid uvula, and cleft palate.
- Orthodontic problems include crowded upper teeth, malocclusion, delayed dentition, ectopic eruption, shovel-shaped incisors, supernumerary teeth, V-shaped maxillary dental arch, bulging alveolar ridges, dentitio tarda, some impaction, partial eruption, idiopathic root resorption, transposition or other aberrations in the position of the tooth germs, and severe crowding.
- Extremities and digits
- The upper limbs are more severely affected than lower limbs. Coalition of distal phalanges and synonychia found in the hands is never present in the feet. The glenohumeral joint and proximal humerus are more severely affected than the pelvic girdle and femur. The elbow is much less severely involved than the proximal portion of the upper limb.
- Syndactyly involves the hands and feet with partial-to-complete fusion of the digits, often involving second, third, and fourth digits. These are often termed mitten hands and sock feet. In severe cases, all digits are fused, with the palm deeply concave and cup-shaped and the sole supinated.
- Hitchhiker posture or radial deviation of short or broad thumbs results from abnormal proximal phalanx.
- Brachydactyly occurs.
- Nailbeds are contiguous (synonychia).
- Some patients have subacromial dimples and elbow dimples during infancy.
- Mobility at the glenohumeral joint is limited with progressive limitation in abduction, forward flexion, and external rotation with growth.
- Limited elbow mobility is common with decreased elbow extension, flexion, pronation, and supination.
- Short humeri are a constant finding beyond infancy.
- Limited genu valga is present in many cases.
- CNS
- Intelligence varies from normal to mental deficiency, although a significant number of patients are mentally retarded. Malformations of the CNS may be responsible for most cases.
- Common CNS malformations include megalencephaly, agenesis of the corpus callosum, malformed limbic structures, variable ventriculomegaly, encephalocele, gyral abnormalities, hypoplastic cerebral white matter, pyramidal tract abnormalities, and heterotopic gray matter. Progressive hydrocephalus is uncommon.
- Papilledema and optic atrophy with loss of vision may be present in cases of subtle increased intracranial pressure.
- Other skeletal and cartilaginous segmentation defects
- Congenital cervical spinal fusion (68%), especially C5-C6
- Aplasia or ankylosis of shoulder, elbow, and hip joints
- Tracheal cartilage anomalies
- Rhizomelia
- Skin
- Hyperhidrosis (common)
- Synonychia
- Brittle nails
- Acneiform lesions (frequent after adolescence)
- Interruption of the eyebrows
- Hypopigmentation
- Hyperkeratosis in the plantar surface
- Paronychial infections (more common in feet than hands and in patients who are institutionalized patients)
- Excessive skin wrinkling of forehead
- Skin dimples at knuckles, shoulders, and elbows
- Cardiovascular (10%)
- Genitourinary (9.6%)
- Polycystic kidneys
- Duplication of renal pelvis
- Hydronephrosis
- Stenosis of bladder neck
- Bicornuate uterus
- Vaginal atresia
- Protuberant labia majora
- Clitoromegaly
- Cryptorchidism
- GI (1.5%)
- Pyloric stenosis
- Esophageal atresia and tracheoesophageal fistula
- Ectopic or imperforate anus
- Partial biliary atresia with agenesis of gallbladder
- Respiratory (1.5%)
- Anomalous tracheal cartilage
- Tracheoesophageal fistula
- Pulmonary aplasia
- Absent right middle lobe of lung
- Absent interlobular lung fissures
Causes
- More than 98% of cases with Apert syndrome are caused by specific missense substitution mutations, involving adjacent amino acids (ie, Ser252Trp, Ser252Phe, Pro253Arg) in the linker between the second and third extracellular immunoglobulin domains of FGFR2, which maps to chromosome bands 10q26. The remaining cases are due to Alu-element insertion mutations in or near exon 9 of FGFR2.
- Most cases are sporadic, resulting from new mutations with a paternal age effect. The incidence of FGFR2 mutations increases exponentially with paternal age, probably due to an increase in the frequency of these mutations and a selective advantage in the male germ line.8,9
- Most new mutations, estimated at 1 per 65,000 live births, imply that germline transversion rates at these 2 positions are currently the highest known in the human genome. The rarity of familial cases can be explained by reduced genetic fitness of individuals because of severe malformations and the presence of mental retardation in many cases.
More on Apert Syndrome |
 Overview: Apert Syndrome Overview: Apert Syndrome |
| Differential Diagnoses & Workup: Apert Syndrome |
| Treatment & Medication: Apert Syndrome |
| Follow-up: Apert Syndrome |













